Running Tensor-cell2cell on your own GPU or on Google Colab's GPU¶
You can run this notebook locally or on Google Colab directly here
Before running this notebook, make sure to have a proper NVIDIA GPU driver (https://www.nvidia.com/Download/index.aspx) as well as the CUDA toolkit (https://developer.nvidia.com/cuda-toolkit) installed.
Then, make sure to create an environment with Pytorch following these instructions to enable CUDA
Citations¶
If you plan using this notebook with your own scRNA-seq data, please cite these references:
- Tensor-cell2cell Tool: Armingol E., Baghdassarian H., Martino C., Perez-Lopez A., Aamodt C., Knight R., Lewis N.E. Context-aware deconvolution of cell-cell communication with Tensor-cell2cell. Nat Commun 13, 3665 (2022). https://doi.org/10.1038/s41467-022-31369-2
- Repository of Ligand-Receptor Pairs used to download the list of LR pairs: Armingol, E., Officer, A., Harismendy, O. et al. Deciphering cell–cell interactions and communication from gene expression. Nat Rev Genet 22, 71–88 (2021). https://doi.org/10.1038/s41576-020-00292-x
- List of Ligand-Receptor Pairs: Jin, S., Guerrero-Juarez, C.F., Zhang, L. et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun 12, 1088 (2021). https://doi.org/10.1038/s41467-021-21246-9
1 - Initial setups¶
Installing necessary libraries¶
# To install a stable version of cell2cell run this:
#!pip install cell2cell
import cell2cell as c2c
import scanpy as sc
import pandas as pd
import numpy as np
from tqdm import tqdm
%matplotlib inline
import warnings
warnings.filterwarnings('ignore')
c2c.__version__
'0.6.4'
2 - Load Data¶
Directories to store data¶
output_folder = './BALF-COVID19/'
c2c.io.directories.create_directory(output_folder)
./BALF-COVID19/ already exists.
Example dataset of COVID-19¶
This is a BALF COVID-19 dataset that consists in 63k immune and epithelial cells in lungs from 3 control, 3 moderate COVID-19, and 6 severe COVID-19 patients.
This dataset was previously published in [1], and this object contains the raw counts for the annotated cell types available in: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145926
Here we obtain an AnnData object containing the raw counts.
References:
[1] Liao, M., Liu, Y., Yuan, J. et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med 26, 842–844 (2020). https://doi.org/10.1038/s41591-020-0901-9
adata = c2c.datasets.balf_covid()
adata
AnnData object with n_obs × n_vars = 63103 × 33538
obs: 'sample', 'sample_new', 'group', 'disease', 'hasnCoV', 'cluster', 'celltype', 'condition'
Ligand-Receptor pairs¶
Different databases of ligand-receptor interactions could be used. We previously created a repository that includes many available DBs (https://github.com/LewisLabUCSD/Ligand-Receptor-Pairs). In this tutorial, we employ the ligand-receptor pairs from CellChat (https://doi.org/10.1038/s41467-021-21246-9), which includes multimeric protein complexes.
lr_pairs = pd.read_csv('https://raw.githubusercontent.com/LewisLabUCSD/Ligand-Receptor-Pairs/master/Human/Human-2020-Jin-LR-pairs.csv')
lr_pairs = lr_pairs.astype(str)
lr_pairs.head(2)
| interaction_name | pathway_name | ligand | receptor | agonist | antagonist | co_A_receptor | co_I_receptor | evidence | annotation | interaction_name_2 | ligand_symbol | receptor_symbol | ligand_ensembl | receptor_ensembl | interaction_symbol | interaction_ensembl | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | TGFB1_TGFBR1_TGFBR2 | TGFb | TGFB1 | TGFbR1_R2 | TGFb agonist | TGFb antagonist | nan | TGFb inhibition receptor | KEGG: hsa04350 | Secreted Signaling | TGFB1 - (TGFBR1+TGFBR2) | TGFB1 | TGFBR1&TGFBR2 | ENSG00000105329 | ENSG00000106799&ENSG00000163513 | TGFB1^TGFBR1&TGFBR2 | ENSG00000105329^ENSG00000106799&ENSG00000163513 |
| 1 | TGFB2_TGFBR1_TGFBR2 | TGFb | TGFB2 | TGFbR1_R2 | TGFb agonist | TGFb antagonist | nan | TGFb inhibition receptor | KEGG: hsa04350 | Secreted Signaling | TGFB2 - (TGFBR1+TGFBR2) | TGFB2 | TGFBR1&TGFBR2 | ENSG00000092969 | ENSG00000106799&ENSG00000163513 | TGFB2^TGFBR1&TGFBR2 | ENSG00000092969^ENSG00000106799&ENSG00000163513 |
Interaction columns contianing the gene names
int_columns = ('ligand_symbol', 'receptor_symbol')
3 - Data Preprocessing¶
Information of samples¶
First, generate a dictionary indicating what condition group is associated to each sample/context
context_dict = adata.obs.set_index('sample')['condition'].sort_values().to_dict()
context_dict
{'C100': 'Control',
'C52': 'Control',
'C51': 'Control',
'C142': 'Moderate COVID-19',
'C141': 'Moderate COVID-19',
'C144': 'Moderate COVID-19',
'C143': 'Severe COVID-19',
'C148': 'Severe COVID-19',
'C149': 'Severe COVID-19',
'C146': 'Severe COVID-19',
'C145': 'Severe COVID-19',
'C152': 'Severe COVID-19'}
Generate list of RNA-seq data (per sample) with single cells aggregated into cell types¶
Here, we convert the single-cell expression levels into cell-type expression levels. A basic context-wise preprocessing is performed here, keeping only genes that are expressed at least in 4 single cells.
Important: A list of aggregated gene expression matrices must be used for building the 4D-communication tensor. Each of the gene expression matrices is for one of the contexts, following the same order as the previous dictionary (context_dict).
Each gene expression is aggregated here across all single cells annotated with the same cell type as the average log1p(CPM) value. Other aggregation methods could be used, please inspect parameter method='average' in the aggregate_single_cell() function. Additionally, other gene expression values could be passed, using other preprocessing approaches such as the non-zero fraction of cells expressing a gene or batch correction methods.
rnaseq_matrices = []
# Iteraty by sample/context
for context in tqdm(context_dict.keys()):
# Obtain metadata for context
meta_context = adata.obs.loc[adata.obs['sample'] == context].copy()
# Single cells in the context
cells = list(meta_context.index)
# Rename index name to identify the barcodes when aggregating expression
meta_context.index.name = 'barcode'
# Subset RNAseq data by the single cells in the sample/context
tmp_adata = adata[cells]
# Keep genes in each sample with at least 4 single cells expressing it
genes = sc.pp.filter_genes(tmp_adata, min_cells=4, inplace=False)[0]
# Convert raw counts into log1p(CPM)
sc.pp.normalize_total(tmp_adata, target_sum=1e6)
sc.pp.log1p(tmp_adata)
# Convert to dataframe and keep filtered genes
tmp_adata = tmp_adata.to_df().loc[:, genes]
# Aggregate gene expression of single cells into cell types
exp_df = c2c.preprocessing.aggregate_single_cells(rnaseq_data=tmp_adata,
metadata=meta_context,
barcode_col='barcode',
celltype_col='celltype',
method='average',
)
rnaseq_matrices.append(exp_df)
100%|███████████████████████████████████████████| 12/12 [00:09<00:00, 1.23it/s]
Preprocessing of LR pairs¶
Generate a dictionary with function info for each LR pairs.
Keys are LIGAND_NAME^RECEPTOR_NAME (this is the same nomenclature that will be used when building the 4D-communication tensor later), and values are the function in the annotation column in the dataframe containing ligand-receptor pairs. Other functional annotations of the LR pairs could be used if available.
ppi_functions = dict()
for idx, row in lr_pairs.iterrows():
ppi_label = row[int_columns[0]] + '^' + row[int_columns[1]]
ppi_functions[ppi_label] = row['annotation']
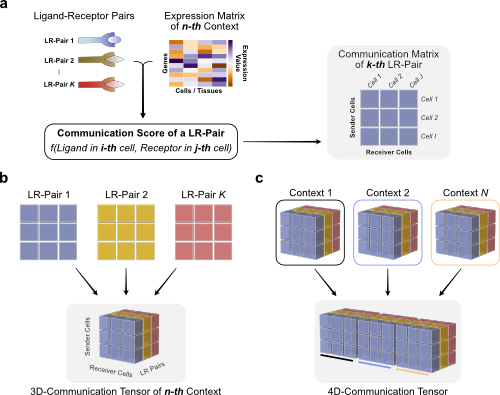
4 - Tensor-cell2cell analysis¶
Build 4D-Communication Tensor Here we use as input the list of gene expression matrices that were aggregated into a cell-type granularity. This list contains the expression matrices of all samples/contexts.
The following functions perform all the steps to build a 4D-communication tensor:
 .
.
The parameter communication_score indicates the scoring function to use.
how='inner' is used to keep only cell types and genes that are across all contexts. Use how='outer' to include all cell types, even if they are not in all samples/contexts. When using the last option, a tensor.mask attribute is created, where zeros indicates what are the missing cell pairs and LR interactions in the given contexts, and ones indicate those that are present. Thus, we can deal with missing values by assigning them a value of zero during the tensor decomposition. We can also control the minimum fraction of samples that must containg a LR pair or a cell type to be included in the tensor, through the parameter outer_fraction.
complex_sep='&' is used to specify that the list of ligand-receptor pairs contains protein complexes and that subunits are separated by '&'. If the list does not have complexes, use complex_sep=None instead.
tensor = c2c.tensor.InteractionTensor(rnaseq_matrices=rnaseq_matrices,
ppi_data=lr_pairs,
context_names=list(context_dict.keys()),
how='outer',
outer_fraction=0.5, # Considers elements in at least 50% of samples
complex_sep='&',
interaction_columns=int_columns,
communication_score='expression_gmean',
)
Getting expression values for protein complexes Building tensor for the provided context
Evaluate some tensor properties¶
Tensor shape
This indicates the number of elements in each tensor dimension: (Contexts, LR pairs, Sender cells, Receiver cells)
tensor.tensor.shape
(12, 807, 10, 10)
Fraction of excluded elements
This represents the fraction of values that are ignored (masked) in the analysis.
tensor.excluded_value_fraction()
0.28569496076001655
Prepare Tensor Metadata¶
To interpret analysis on the tensor, we can assign groups to each sample/context, and to every elements in the other dimensions (LR pairs and cells).
We can generate respective dictionaries manually or automatically from DBs.
meta_tensor = c2c.tensor.generate_tensor_metadata(interaction_tensor=tensor,
metadata_dicts=[context_dict, ppi_functions, None, None],
fill_with_order_elements=True
)
Run Tensor-cell2cell¶
Here we use the tf_optimization='regular', which is faster but generates less robust results. We recommend using tf_optimization='robust, which takes longer to run.
tensor2 = c2c.analysis.run_tensor_cell2cell_pipeline(tensor,
meta_tensor,
copy_tensor=True, # Whether to output a new tensor or modifying the original
rank=None, # Number of factors to perform the factorization. If None, it is automatically determined by an elbow analysis
tf_optimization='regular', # To define how robust we want the analysis to be.
random_state=888, # Random seed for reproducibility
backend='pytorch', # This enables a banckend that supports using a GPU.
device='cuda', # Device to use. If using GPU and PyTorch, use 'cuda'. For CPU use 'cpu'
elbow_metric='error', # Metric to use in the elbow analysis.
smooth_elbow=False, # Whether smoothing the metric of the elbow analysis.
upper_rank=25, # Max number of factors to try in the elbow analysis
tf_init='random', # Initialization method of the tensor factorization
tf_svd='numpy_svd', # Type of SVD to use if the initialization is 'svd'
cmaps=None, # Color palettes to use in color each of the dimensions. Must be a list of palettes.
sample_col='Element', # Columns containing the elements in the tensor metadata
group_col='Category', # Columns containing the major groups in the tensor metadata
fig_fontsize=14, # Fontsize of the figures generated
output_folder=output_folder, # Whether to save the figures and loadings in files. If so, a folder pathname must be passed
output_fig=True, # Whether to output the figures. If False, figures won't be saved a files if a folder was passed in output_folder.
fig_format='pdf', # File format of the figures.
)
Running Elbow Analysis
0%| | 0/25 [00:00<?, ?it/s]
The rank at the elbow is: 10 Running Tensor Factorization Generating Outputs Loadings of the tensor factorization were successfully saved into ./BALF-COVID19//Loadings.xlsx


Top genes in each factor
for i in range(tensor2.rank):
print(tensor2.get_top_factor_elements('Ligand-Receptor Pairs', 'Factor {}'.format(i+1), 10))
print('')
CD99^CD99 0.396975 MIF^CD74&CXCR4 0.368345 MIF^CD74&CD44 0.348051 LGALS9^PTPRC 0.319667 TNFSF13B^TNFRSF13B 0.244478 LGALS9^CD44 0.239305 APP^CD74 0.226378 SELPLG^SELL 0.223956 PTPRC^CD22 0.206616 CXCL10^CXCR3 0.174164 Name: Factor 1, dtype: float32 MIF^CD74&CD44 0.607854 LGALS9^CD44 0.450888 CD99^CD99 0.402892 LGALS9^PTPRC 0.260705 LGALS9^HAVCR2 0.258238 MIF^CD74&CXCR4 0.166606 APP^CD74 0.142985 COL9A2^CD44 0.112663 CD22^PTPRC 0.103688 NAMPT^ITGA5&ITGB1 0.098853 Name: Factor 2, dtype: float32 CCL8^CCR1 0.328729 SPP1^CD44 0.318466 CCL3L1^CCR1 0.311767 CCL3^CCR1 0.311655 CCL7^CCR1 0.280976 ANXA1^FPR1 0.240010 MIF^CD74&CXCR4 0.191990 NAMPT^ITGA5&ITGB1 0.188227 MIF^CD74&CD44 0.179285 SPP1^ITGA5&ITGB1 0.165535 Name: Factor 3, dtype: float32 CCL5^CCR1 0.405997 CD99^PILRA 0.308518 ANXA1^FPR1 0.298691 PTPRC^MRC1 0.209846 CCL5^CCR5 0.194877 CD99^CD99 0.193112 ANXA1^FPR2 0.189305 MIF^CD74&CXCR4 0.182442 SELPLG^SELL 0.169664 ITGAL&ITGB2^ICAM1 0.165196 Name: Factor 4, dtype: float32 MDK^NCL 0.379103 APP^CD74 0.365288 LAMC2^CD44 0.290392 LAMB3^CD44 0.283392 LAMA5^CD44 0.221012 PTN^NCL 0.194058 GDF15^TGFBR2 0.178591 LAMB2^CD44 0.167368 LAMA3^CD44 0.153982 COL4A5^CD44 0.147150 Name: Factor 5, dtype: float32 PTPRC^MRC1 0.392890 FN1^CD44 0.374219 MIF^CD74&CD44 0.336574 RETN^CAP1 0.302886 LGALS9^CD44 0.274760 GRN^SORT1 0.239909 HLA-DMA^CD4 0.239026 LGALS9^PTPRC 0.222783 PECAM1^PECAM1 0.168159 RETN^TLR4 0.152095 Name: Factor 6, dtype: float32 APP^CD74 0.463218 HLA-DMA^CD4 0.393801 LGALS9^PTPRC 0.250710 HLA-DOA^CD4 0.226411 LGALS9^HAVCR2 0.209939 GRN^SORT1 0.196472 IL16^CD4 0.183482 CD99^PILRA 0.183337 ICAM1^SPN 0.174727 PECAM1^PECAM1 0.164378 Name: Factor 7, dtype: float32 CD99^CD99 0.279922 NAMPT^INSR 0.274649 SEMA4D^PLXNB2 0.239051 GRN^SORT1 0.223482 SEMA4D^PLXNB1 0.199096 SELL^PODXL 0.187005 TNFSF10^TNFRSF10A 0.169820 AREG^EGFR 0.156962 TGFB1^ACVR1B&TGFBR2 0.152839 AREG^EGFR&ERBB2 0.152592 Name: Factor 8, dtype: float32 CD99^CD99 0.344679 LGALS9^PTPRC 0.341739 MIF^CD74&CD44 0.220866 MIF^CD74&CXCR4 0.212046 CXCL16^CXCR6 0.210066 CXCL10^CXCR3 0.209790 CD48^CD244 0.207834 CCL4^CCR5 0.188202 CCL5^CCR5 0.175231 ALCAM^CD6 0.174957 Name: Factor 9, dtype: float32 RETN^CAP1 0.394428 FN1^CD44 0.363788 MDK^NCL 0.291849 SIGLEC1^SPN 0.274590 LGALS9^CD44 0.228481 LGALS9^PTPRC 0.193877 THBS1^CD47 0.177242 CCL3^CCR1 0.141885 CCL8^CCR1 0.139728 LGALS9^HAVCR2 0.126326 Name: Factor 10, dtype: float32
5 - Export objects¶
Beyond the generated results, we can export the objects here for using them in posterior analyses.
# Export Tensor after decomposition
c2c.io.export_variable_with_pickle(tensor, output_folder + 'Tensor.pkl')
./BALF-COVID19/Tensor.pkl was correctly saved.
# Export Tensor Metadata
c2c.io.export_variable_with_pickle(meta_tensor, output_folder + 'Tensor-Metadata.pkl')
./BALF-COVID19/Tensor-Metadata.pkl was correctly saved.